Invented by Bahram Ghaffarzadeh Kermani, Helmy Eltoukhy, Guardant Health Inc
Cell-free tumor nucleic acids sequencing involves the analysis of ctDNA and ctRNA, which are released into the bloodstream by tumor cells. These nucleic acids carry genetic information that can be used to identify and monitor cancer-related mutations. However, the detection of single nucleotide somatic variants (SNVs) in ctDNA and ctRNA is a complex task due to the low abundance and high noise levels of these molecules.
Machine learning algorithms offer a solution to this challenge by leveraging computational models to analyze large-scale genomic data and identify patterns associated with SNVs. These algorithms can learn from vast amounts of training data, allowing them to detect even subtle genetic variations that might be missed by traditional sequencing methods.
The market for machine learning in single nucleotide somatic variant detection is driven by several factors. Firstly, the increasing prevalence of cancer worldwide has created a demand for more accurate and efficient diagnostic tools. Machine learning algorithms can provide highly sensitive and specific SNV detection, enabling early cancer detection and personalized treatment strategies.
Secondly, the advancements in genomic sequencing technologies have made it possible to generate vast amounts of genomic data at a lower cost. Machine learning algorithms can effectively handle this big data and extract meaningful insights, facilitating the identification of SNVs in ctDNA and ctRNA.
Furthermore, the integration of machine learning algorithms with existing bioinformatics pipelines and sequencing platforms has become more seamless, making it easier for researchers and clinicians to adopt these technologies. This has led to a growing number of collaborations between machine learning companies and genomics research institutions, further driving the market growth.
Several key players are actively involved in the development and commercialization of machine learning tools for SNV detection in cell-free tumor nucleic acids sequencing applications. These companies are investing in research and development to improve the accuracy and efficiency of their algorithms. They are also focusing on developing user-friendly software platforms that can be easily integrated into existing workflows.
In addition to the commercial sector, academic and research institutions are also contributing to the growth of this market. Many research groups are exploring novel machine learning approaches to improve SNV detection and develop predictive models for cancer prognosis and treatment response.
However, there are still challenges that need to be addressed in this market. The interpretation of SNVs detected in ctDNA and ctRNA remains a complex task, requiring further research and validation. Additionally, the integration of machine learning algorithms into clinical practice requires regulatory approvals and validation studies to ensure their reliability and safety.
In conclusion, the market for machine learning in single nucleotide somatic variant detection in cell-free tumor nucleic acids sequencing applications is expanding rapidly. This technology has the potential to revolutionize cancer diagnosis and treatment by providing highly accurate and personalized genomic information. With ongoing advancements in machine learning algorithms and genomic sequencing technologies, we can expect further growth and innovation in this field.

The Guardant Health Inc invention works as follows
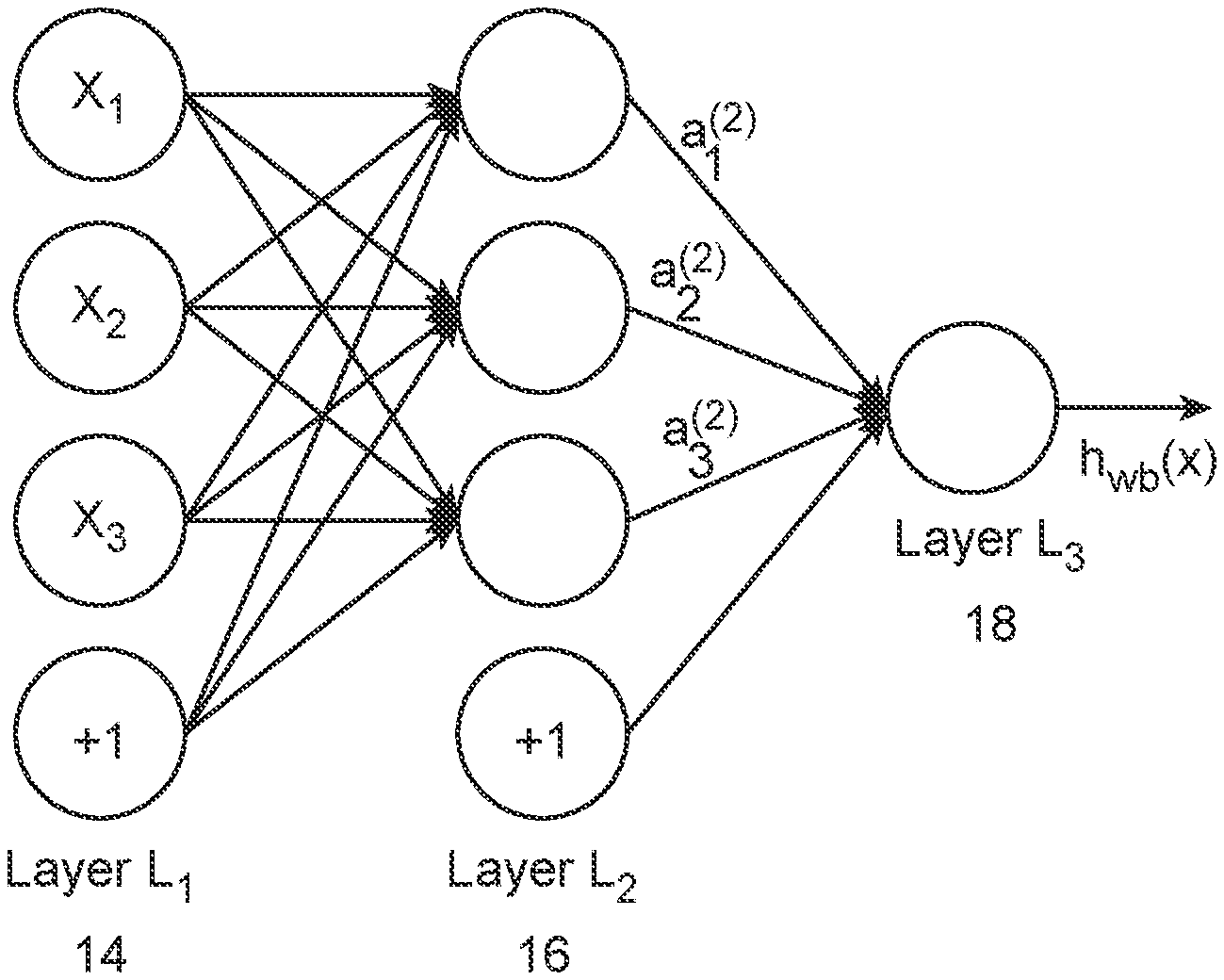
The following systems and methods are described to detect single nucleotide variants (SNVs), from somatic origins, in a cell free biological sample from a subject. These include generating training data and class labels, creating a machine-learning unit in computer memory that has one output per adenine, cytosine, guanine, and thymine calls, training the machine-learning unit, and then applying the machine-learning unit to detect SNVs, from somatic origins, in

Background for Machine learning for single nucleotide somatic variant detection in cell free tumor nucleic acids sequencing applications
Single nucleotide variations (SNVs) detection is an important step in the typical analysis pipeline of re-sequencing. The detection of (or determination of) single-base variations between a newly created sequence and a standard reference sequence is what it refers to. There are also other types of variations that can occur between a sample’s DNA sequence and the reference sequence. These variations include: 1) indels (e.g. deletions or insertions), 2) copy number variations (CNVs), including changes that can involve very long stretches of nucleotides (e.g. thousands or millions), and 3) chromosomal rearrangements such as gene fusions. Indel is a term used to describe either an insertion (or Indel) or deletion (or Indels) at a particular location. The detection of the two types of variants mentioned above is more difficult than that of SNVs. However, the present disclosure can be applied to both of them, as those with the relevant knowledge will understand.
Variant detection is followed by a mapping or aligning step in the pipeline. This includes SNV detection and indel detection as well as SV or CNV. The mapping or alignment operation is the process by which the sequencing reads from the original sequence are mapped onto the reference sequence. The short sequencing reads and the repeated regions of the long reference sequence (e.g. the human genome is about?3 billion nucleotides) make it difficult to find the exact position where the read should be mapped. The methods of genome mapping, which are well-known to experts in the field, are not covered here.
One of the reasons SNV detection can be difficult with next-generation sequencing approaches (NGS), is that the error rate produced by the conventional NGS technology (e.g. Illumina technology) has been widely believed to range between 0.1% and 1%. This is a zero to one order magnitude higher than SNV rate. SNVs are both reflected in differences between the sequence data and the reference. The ‘noise’ is the difference between the sequencing data and reference sequence. The ‘noise’ (e.g. sequencing error) can be up to an order of magnitude greater than the signal? “(e.g. real SNVs).
The present disclosure describes methods and systems to detect single-nucleotide variants (SNVs), from somatic sources, in a cellless biological sample from a subject. This could be in a mixture nucleic acids from both somatic and germline origins.
The following systems and methods are described to detect single nucleotide variants (SNVs), from somatic origins, in a sample of cell-free tissue of a person. They include generating training data and class labels, forming a machine-learning unit that has one output each for adenine, cytosine, guanine, and thymine base calls; training the unit with biological samples, and then applying the unit to detect SNVs, from somatic origins, in
Advantages” of the system can include any or all of the following. The system is capable of handling a wide range of input features for SNV detections. The GC content (which can be very informative) is one example. The system is highly scalable. The system doesn’t rely on hard thresholds for the number of molecules, which is helpful in scaling when there are variable coverage. The system is able to make accurate decisions even when there are deviations (either higher or lower) from the nominal value. The system offers optimal SNV detection?in contrast, heuristic methods do not guarantee optimal detection. The system offers probabilistic quantification, with a score of quality that can be used globally in an downstream probabilistic method (e.g. Bayesian).
The following detailed description will make it clear to those in the art that the disclosure has additional aspects and benefits. Only illustrative examples are described and shown. The present disclosure can be adapted to other embodiments and details, without departing from its disclosure. The drawings and descriptions are therefore to be considered as indicative and not restrictive.
INCORPORATION BY RESEARCH
All publications, patents and patent applications mentioned herein are herein incorporated as if each publication, patent, and/or patent application were specifically and individually indicated that they would be incorporated by refer.
It will be clear to those who are skilled in the art, that the various embodiments described and shown herein are only examples. Those skilled in the arts may make a number of changes and substitutions without compromising the invention. “It should be understood that alternative embodiments to the invention described in this document may be used.

The “cell-free deoxyribonucleic (DNA)” from a cancer patient is a mixture that contains both germline DNA and DNA from cancer cells, which contain somatic mutations. Base calls are made at genomic base positions (hereafter, “base position”) when such a mixture of DNA is sequenced. When the cancer cells are mutated, the sequence reads will contain a mixture of base calls from both the germline and somatic DNA. There is also the possibility of errors in sequencing. For example, 1,000 reads of a cancer patient’s DNA may result in A=988, G=1, T=2, and C=9. In this case, the user could call C’s presence at the genomic position base in the sample and call it at 1%. This allows for the determination of a number of genomic bases in a sample. The presence of a base or bases at each genomic base, their relative frequency, and/or probability can be called.
Machine learning can be used to create models that predict the presence of bases at genomic base positions in samples containing mixed DNA (e.g. germline DNA and cellular DNA) more accurately than a heuristic approach. Optionally, the model can also provide a confidence rating of the call. These models can be created by providing machine learning units with data that has the output known in advance. “An output where it is known that 98% of bases at a genomic position are A, and 1% are B.
This type of training set is available in the following form. Sequencing of cell-free DNA can be done from multiple samples that are presumably homogenous. Cell-free DNA can be obtained from healthy or non-cancer people, who don’t have somatic mutations in their diseased cells. This gives a sequence of bases at each genomic position that is examined. The base should be the same in all the molecules that are homozygous for that locus. This can produce for each sample a vector that indicates, at each genomic position, the counts of the bases at the genomic position.
The polymorphism in the human population at any genomic position is around 0.1%.” In any given sample, it is expected that 0.1% of base positions will harbor variants. This would mean, for a panel of 160.000 bases, approximately 160 variants. In other words, in a sample set of 1,000, in worst-case scenario, you might expect to see a single nucleotide variant (SNP) at a particular base position in one of the samples.
The method can now proceed either in vitro or in silico. Both methods involve reading mixtures of samples.
All mixtures are sequenced. A vector is produced that includes data for each mixture indicating the read or molecule counts (or %) at each base in each mixture. The vector can also include other features, such as GC at a particular base position, entropy or detection of reads on both strands. This is the training set.
The resulting training set is provided to a machine-learning unit such as a neural net or support vector machine. The machine learning unit can generate a classifier model using the training set to classify a sample according to the base identity in one or more positions. Also known as “calling” A base. The model can use information from any part a test vector. It may include information from tally-vectors at other base positions, either proximal to or distal from the test position, or information that is not sequence read.
Click here to view the patent on Google Patents.